Current Research
RNA Metabolism
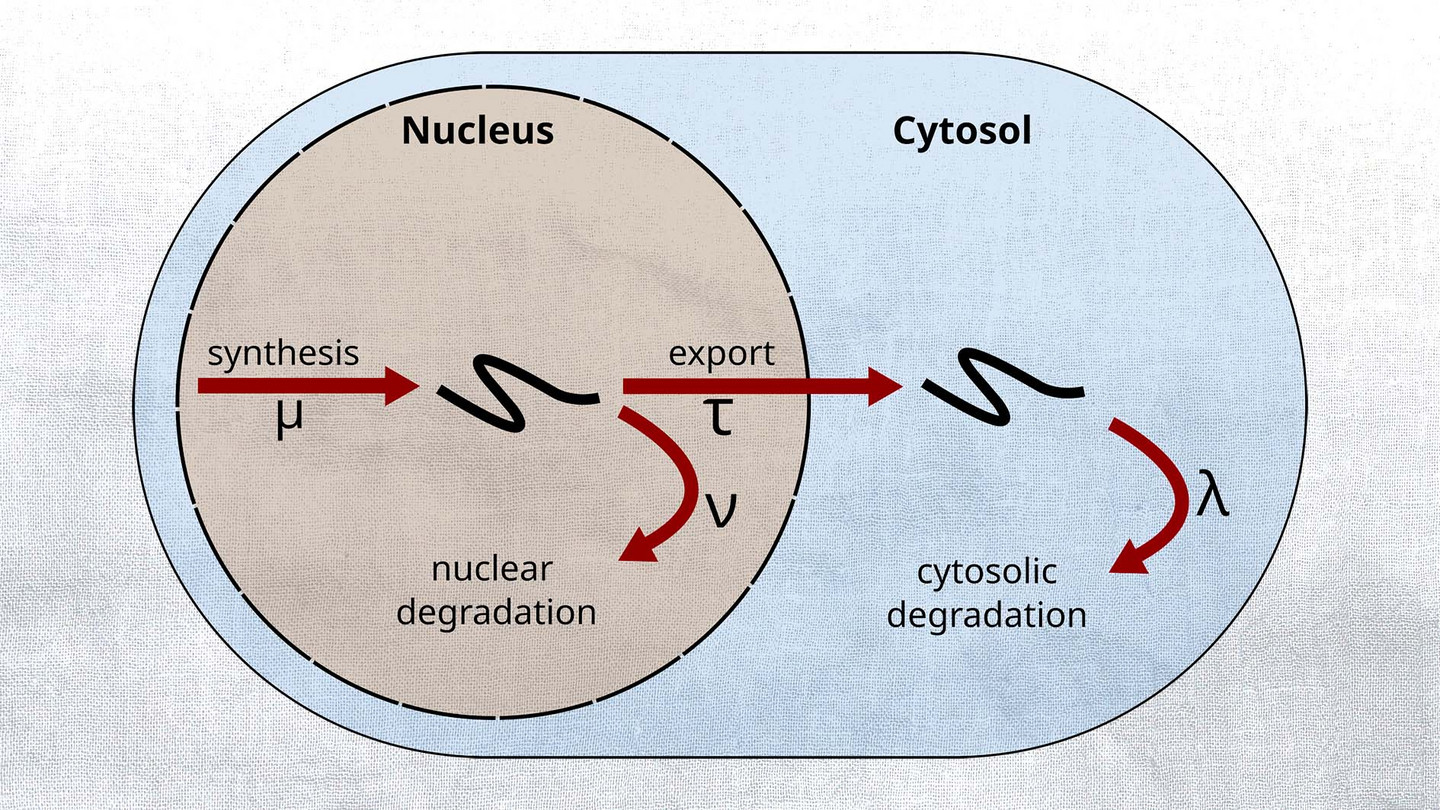
Gene expression regulation is of vital importance to the functionality of the cell. Coordinated expression changes are crucial to the cell cycle, environmental adaptations, and cell differentiation. In the related projects, we develop a new biochemical-bioinformatics approach for the measurement of the kinetic parameters of mRNA metabolism (i.e., RNA synthesis rate, nuclear and cytosolic degradation, and RNA export rate). Using RNA metabolic labelling in combination with deep sequencing, we can distinguish transcripts that have been synthesized after the application of the labelling pulse from pre-existing ones. With this technique, we analyze time series data for nuclear and cytosolic RNA. This allows us to identify RNA metabolic parameters on a genome-wide scale.
Müller 2024/Baar 2022/Schwalb 2016/Eser 2014/Sun 2012/Zacher 2011
Single-Cell Omics
Cell-fate decisions typically involve epigenetic and transcriptional, as well as proteomic changes. Recent technological developments capture one or at most two of those modalities on a single-cell level. Depending on the method, the tissue sample is either lyzed or can be processed as a whole, thus preserving the spatial relations between individual cells (spatial profiling). However, the statistically sound data integration poses severe challenges. Based on multi-omics data, our group constructs novel representations of cell states that do not necessarily rely on prior dimensionality reduction as, e.g., done by neural networks.
Rosendahl 2023/Nikopoulou 2023/Hussainy 2022/Failmezger 2021/Schlusche 2021
Statistics & Algorithms for High-Dimensional Data
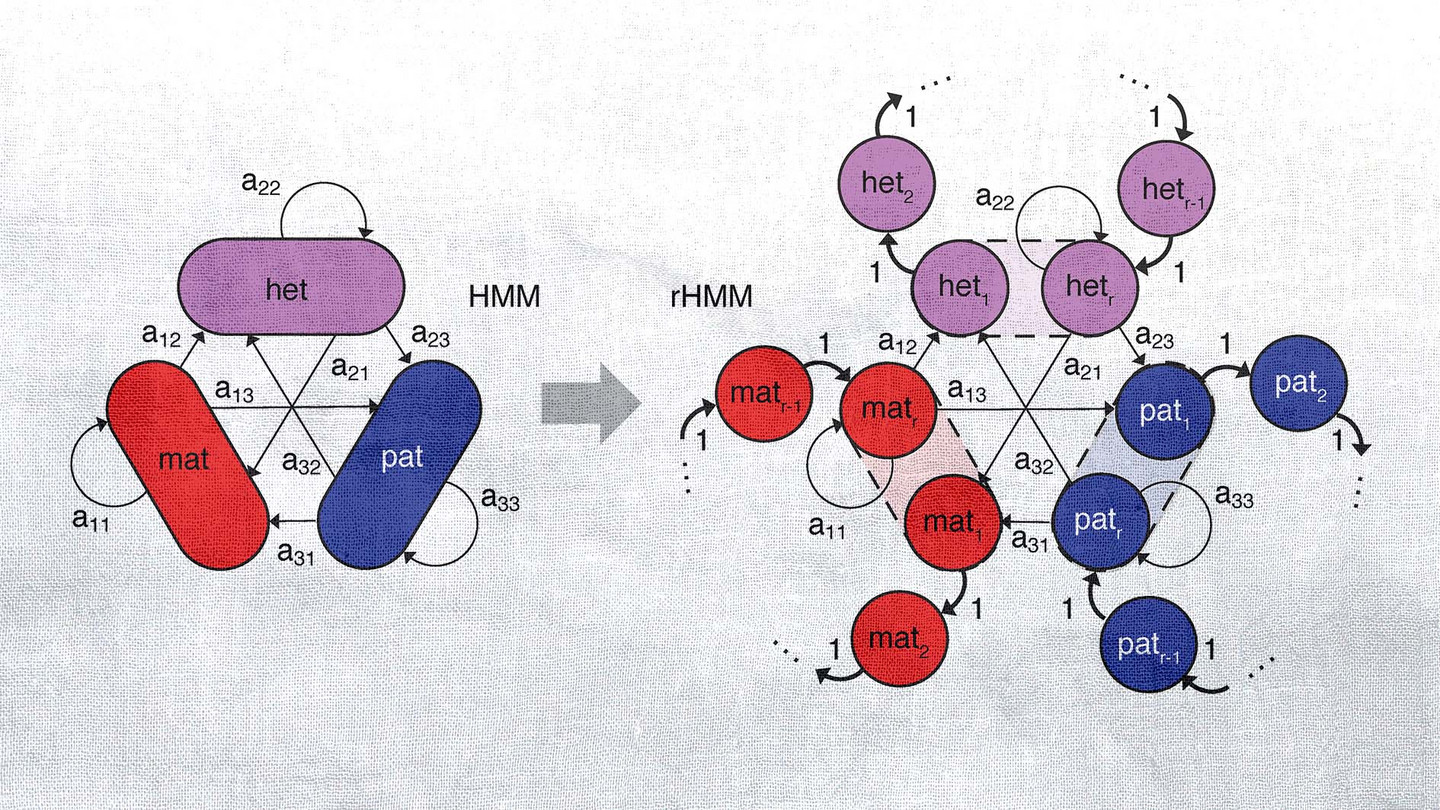
Epigenomics data (e.g., ChIP-seq or Cut&Run) is extremely powerful in characterising the accessibility and activity of the genome. Yet, in theory, the signal-to-noise ratio per base pair is prohibitively low. This treasure trove of data can only be salvaged if the information contained in neighbouring positions is brought together skillfully. To this end, we have developed a number of algorithms capable of processing sequential data. By applying these, we were able to dissect the RNA Polymerase II transcription cycle into distinct phases. Furthermore, we could assemble high-precision maps of cell-type specific, active promoters and enhancers.
Kleinenkuhnen 2023/Campos-Martin 2022/Failmezger 2018/Stricker 2017/Glas 2015/Zacher 2014